Канаван ауруы: генетикасы, патогенезі, клиникалық белгілері және диагностикасы


ҚАЗАҚСТАН РЕСПУБЛИКАСЫ АУЫЛ ШАРУАШЫЛЫҒЫ МИНИСТРЛІГІ
Жәңгір хан атындағы Батыс Қазақстан аграрлық-техникалық университеті
«Жұқпалы емес аурулар және морфология» кафедрасы
Орындаған:
ВМ-23 тобының студенті
Каримова Г. З.
Тексерген: а. о Батыргалиев Е. А.

Кэнэвэн синдромы - мидың жүйке жасушаларының үдемелі зақымдануымен сипатталатын тұқым қуалайтын ауру. Бұл ауру генетикалық негізделген лейкодистрофиялар тобына жатады. Лейкодистрофияда жүйке талшықтарының миелинді қабығы бұзылады. Ауру негізінен еврей-ашкеназдардың арасында таралған. Сонымен қатар кэнэвэен синдромы аспартоацилаза генінің мутациясымен байланысты сирек кездесетін ауто-рецессивті ауруы болып табылады. Бұл ферменттің тапшылығының нәтижесі орталық жүйке жүйесінде N-ацетил-L-аспарагин қышқылының жинақталуы және ацетат тапшылығы болып табылады, бұл бас және жұлын миының ақ және сұр затының кеуекті дегенерациясының қалыптасуына әкеледі.

Канаван ауруы алғаш рет зерттелген және 1931 жылы американдық дәрігер сипатталған. Өз атауын тұқым қуалайтын патология өзінің алғашқы ашушысы - Миртель Канаван алды. Кейінірек 1991 жылы клиницистер геномдық бірлікті бөліп алды, оған патологиялық тұқым қуалаушылықтың қалыптасуы мен дамуы толық байланысты. Ғылыми жетістіктердің арқасында, бүгінгі күні кэнэвэн ауруын ұрықтың құрсақішілік даму кезеңінде манифестациялауға болады, әсіресе оң молекулалық-генетикалық зерттеулер кезінде. Ауру кейбір өлшемдер бойынша ерекшеленеді.
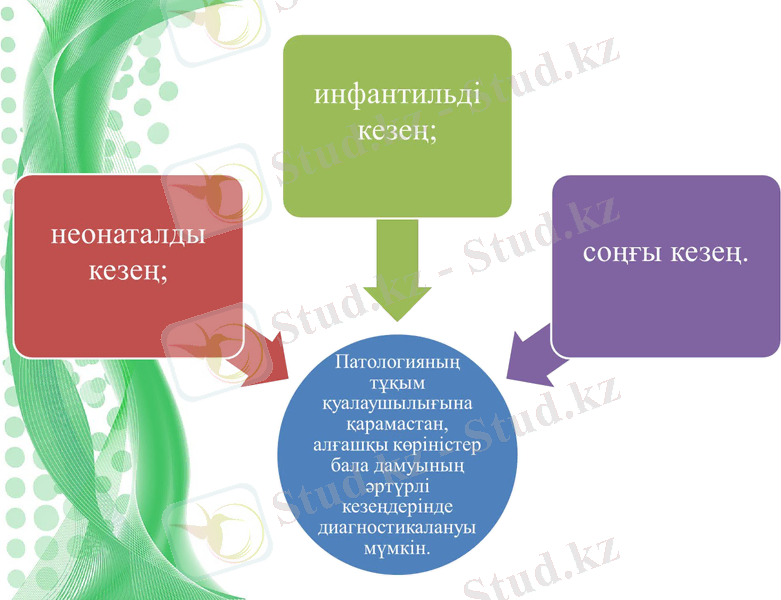

Қазіргі неврологияда ауру миелиндегі липидтік алмасудың бұзылуының салдарынан болады, ол 75% - ға ақ ми затының құрамын анықтайды. Кейбір симптомдар бойынша атап өтуге болатын патологиялық бұзылулар әртүрлі кезеңдерде шыңына жетеді. Инфантильді кезеңде Кэнэвэн ауруының пайда болуы кең таралған.
Балаларда Кэнэвэн ауруының тұқым қуалау түрі-аутосомно-рецессивті, ақаулы геннің ата-анасының екеуінен де берілуі болған кезде. Бұл ретте, егер анасы да, әкесі де аномальды мутацияларды тасушы болып табылса, онда баланың нейровегетативті-тұқым қуалаушылық бұзылулары бар тууының нақты тәуекелдері 30% - ды құрайды. Бүгін әлеуетті ата-аналардың генетикалық панелін зерттеу түрлі гендік мутацияларды анықтауға мүмкіндік береді. Әдетте анамнезінде Канаван ауруы бар 17 хромосома патологиялық болып табылады.
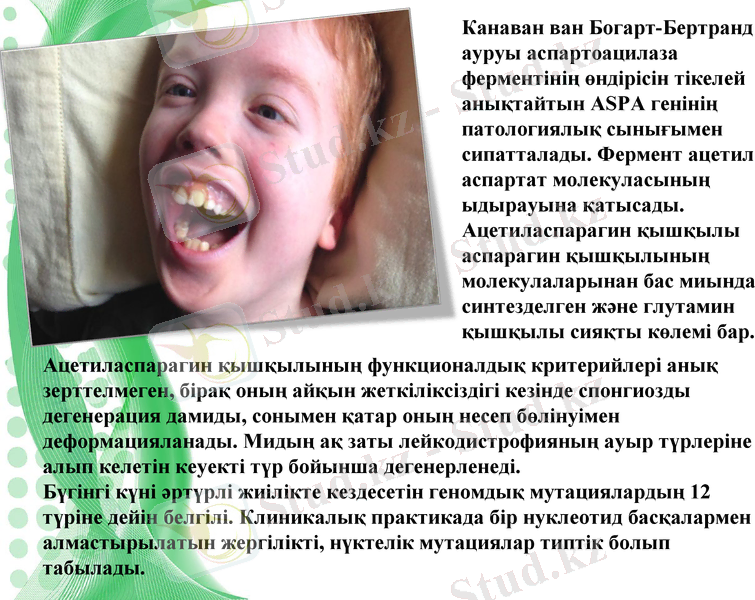
Канаван ван Богарт-Бертранд ауруы аспартоацилаза ферментінің өндірісін тікелей анықтайтын ASPA генінің патологиялық сынығымен сипатталады. Фермент ацетил аспартат молекуласының ыдырауына қатысады.
Ацетиласпарагин қышқылы аспарагин қышқылының молекулаларынан бас миында синтезделген және глутамин қышқылы сияқты көлемі бар.
Ацетиласпарагин қышқылының функционалдық критерийлері анық зерттелмеген, бірақ оның айқын жеткіліксіздігі кезінде спонгиозды дегенерация дамиды, сонымен қатар оның несеп бөлінуімен деформацияланады. Мидың ақ заты лейкодистрофияның ауыр түрлеріне алып келетін кеуекті түр бойынша дегенерленеді.
Бүгінгі күні әртүрлі жиілікте кездесетін геномдық мутациялардың 12 түріне дейін белгілі. Клиникалық практикада бір нуклеотид басқалармен алмастырылатын жергілікті, нүктелік мутациялар типтік болып табылады.

Канаван ауруының белгілері әдетте 3-тен 6 айға дейінгі балаларда пайда болады. Симптомдарға мыналар жатады: баланың дамуының кідіруі, бастың ұлғаюы (макроцефалия), бұлшық ет тонусының жоғалуы (гипотония), баланы емізудегі елеулі проблемалар, құрысулар, гиперкинез, бас сүйек жүйкелерінің зақымдану белгілері, адинамия.
Канаван ауруын емдеу қолдау сипатында, мидың миелинді компоненттерін толығымен қалпына келтіре алмайды. Бүгінгі таңда жатырішілік даму кезеңінде және туғаннан кейін Канаван ауруынан құтылуға мүмкіндік беретін белгілі бір әдістеме жоқ. Медицина қызметкерлерінің жалғыз нәрсе-метаболикалық терапияны тағайындау арқылы аурудың прогрессиясын баяулатады. Емдеу миелиннің бұзылуын тоқтатуға мүмкіндік беретін дәрі-дәрмектерді өмір бойы қабылдауды талап етеді.
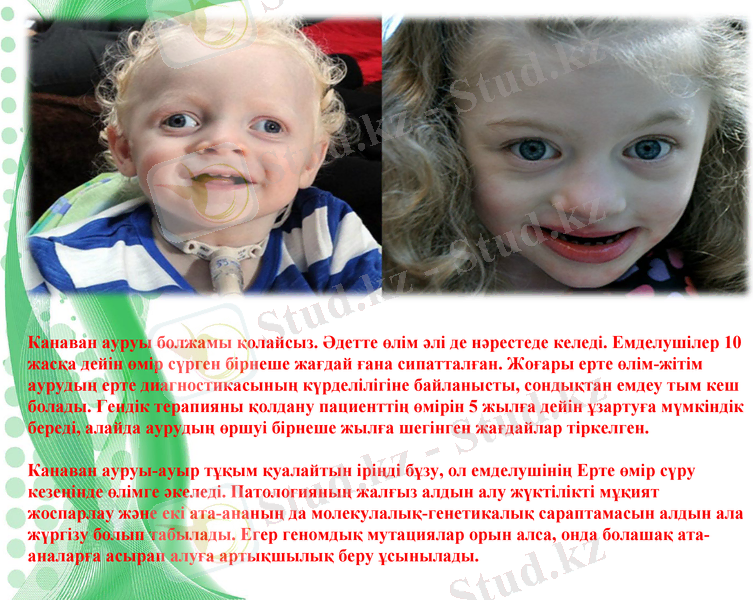
Канаван ауруы болжамы қолайсыз. Әдетте өлім әлі де нәрестеде келеді. Емделушілер 10 жасқа дейін өмір сүрген бірнеше жағдай ғана сипатталған. Жоғары ерте өлім-жітім аурудың ерте диагностикасының күрделілігіне байланысты, сондықтан емдеу тым кеш болады. Гендік терапияны қолдану пациенттің өмірін 5 жылға дейін ұзартуға мүмкіндік береді, алайда аурудың өршуі бірнеше жылға шегінген жағдайлар тіркелген.
Канаван ауруы-ауыр тұқым қуалайтын іріңді бұзу, ол емделушінің Ерте өмір сүру кезеңінде өлімге әкеледі. Патологияның жалғыз алдын алу жүктілікті мұқият жоспарлау және екі ата-ананың да молекулалық-генетикалық сараптамасын алдын ала жүргізу болып табылады. Егер геномдық мутациялар орын алса, онда болашақ ата-аналарға асырап алуға артықшылық беру ұсынылады.
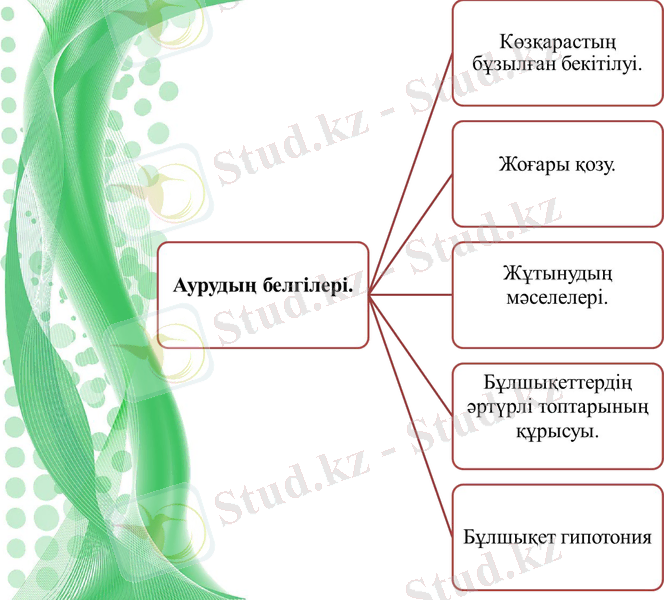
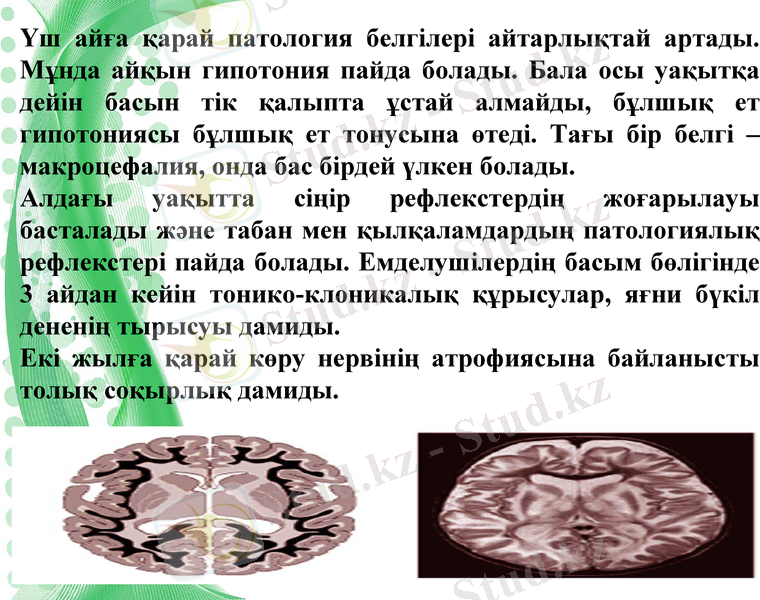
Үш айға қарай патология белгілері айтарлықтай артады. Мұнда айқын гипотония пайда болады. Бала осы уақытқа дейін басын тік қалыпта ұстай алмайды, бұлшық ет гипотониясы бұлшық ет тонусына өтеді. Тағы бір белгі - макроцефалия, онда бас бірдей үлкен болады.
Алдағы уақытта сіңір рефлекстердің жоғарылауы басталады және табан мен қылқаламдардың патологиялық рефлекстері пайда болады. Емделушілердің басым бөлігінде 3 айдан кейін тонико-клоникалық құрысулар, яғни бүкіл дененің тырысуы дамиды.
Екі жылға қарай көру нервінің атрофиясына байланысты толық соқырлық дамиды.

Профилактикасы
Алдын алу ретінде міндетті түрде ата-аналарға бала туар алдында медициналық-генетикалық кеңес беру жүргізіледі. Егер отбасында немесе босануда осы патологиясы бар баланың туу жағдайлары болса, мұндай кеңес әсіресе қажет. Бұл ретте науқас баланың даму қаупі анықталады, сондай-ақ қажет болған жағдайда жүктілік кезінде Кэнэвэн ауруы бар баланың ұрықтығын анықтауға көмектесетін барлық зерттеулер жүргізіледі.
Жүктілік кезінде Диагностика бірнеше жолмен жүргізіледі. Бұл, мысалы, хориалды ворсинді зерттеу әдісі арқылы баланың гендік жиынтығын тестілеу. Сондай-ақ N-ацетил-аспаргин қышқылының жоғары концентрациясы табылуы мүмкін жеміс жанындағы суларға талдау жүргізілуі мүмкін.

Аурудың диагностикасы.
Алдын алу ретінде міндетті түрде ата-аналарға бала туар алдында медициналық-генетикалық кеңес беру жүргізіледі. Егер отбасында немесе босануда осы патологиясы бар баланың туу жағдайлары болса, мұндай кеңес әсіресе қажет. Бұл ретте науқас баланың даму қаупі анықталады, сондай-ақ қажет болған жағдайда жүктілік кезінде Кэнэвэн ауруы бар баланың ұрықтығын анықтауға көмектесетін барлық зерттеулер жүргізіледі.
Жүктілік кезінде Диагностика бірнеше жолмен жүргізіледі. Бұл, мысалы, хориалды ворсинді зерттеу әдісі арқылы баланың гендік жиынтығын тестілеу. Сондай-ақ N-ацетил-аспаргин қышқылының жоғары концентрациясы табылуы мүмкін жеміс жанындағы суларға талдау жүргізілуі мүмкін.

Тарихы.
- Іс жүргізу
- Автоматтандыру, Техника
- Алғашқы әскери дайындық
- Астрономия
- Ауыл шаруашылығы
- Банк ісі
- Бизнесті бағалау
- Биология
- Бухгалтерлік іс
- Валеология
- Ветеринария
- География
- Геология, Геофизика, Геодезия
- Дін
- Ет, сүт, шарап өнімдері
- Жалпы тарих
- Жер кадастрі, Жылжымайтын мүлік
- Журналистика
- Информатика
- Кеден ісі
- Маркетинг
- Математика, Геометрия
- Медицина
- Мемлекеттік басқару
- Менеджмент
- Мұнай, Газ
- Мұрағат ісі
- Мәдениеттану
- ОБЖ (Основы безопасности жизнедеятельности)
- Педагогика
- Полиграфия
- Психология
- Салық
- Саясаттану
- Сақтандыру
- Сертификаттау, стандарттау
- Социология, Демография
- Спорт
- Статистика
- Тілтану, Филология
- Тарихи тұлғалар
- Тау-кен ісі
- Транспорт
- Туризм
- Физика
- Философия
- Халықаралық қатынастар
- Химия
- Экология, Қоршаған ортаны қорғау
- Экономика
- Экономикалық география
- Электротехника
- Қазақстан тарихы
- Қаржы
- Құрылыс
- Құқық, Криминалистика
- Әдебиет
- Өнер, музыка
- Өнеркәсіп, Өндіріс
Қазақ тілінде жазылған рефераттар, курстық жұмыстар, дипломдық жұмыстар бойынша біздің қор #1 болып табылады.

Ақпарат
Қосымша
Email: info@stud.kz