Гемоглобинопатиялар: талассемия мен орақ тәрізді анемияның клиникасы, диагностикасы және емі


Орындаған: Алжан Толғанай
Группа: 141 ЖМ
Тексерген: Сманова Гульмира Бамадиновна
«Астана медицина университеті» КеАҚ «Молекулалық биология және медициналық генетика»
БӨЖ
Тұқым қуалайтын аурулар. Гемоглобинопатия

Жоспар
І. Кіріспе
ІІ. Негізгі бөлім
2. 1. Талассемия
Клиникалық көрінісі
Диагностикасы
Емі
2. 2. Орақ жасушалы анемия
Этиологиясы және патогенезі
Клиникалық көрінісі
Емі
ІІІ. Қорытынды
IV. Пайдаланылған әдебиеттер

Кіріспе
Қазіргі кезде тұқым қуалайтын аурулар өте көп таралуда. Олардың көбісі туыстық некеден немесе адамның ата-анасынан берілетін ауруллар болып табылады. Бұл аурулардың көбісінде емі жок болып келеді, яғни адам өмірінің соңына дейін осы аурумен ауырады. Менің ойымша, егер ғылымды, медицинаны дамытса бұл аурулардың кейбіреулерін емдеуге жол табуға болады деген ойдамын. Алдымен Гемоглобинопатиялар туралы айтпай тұрып біз тұқым қуалайтын аурулардың анықтамасы мен классификациясы жайлы білуіміз керек. Осыған тоқталатын болсақ:
Тұқым қуалайтын аурулар - ата-аналарынан ұрпақтарына берілетін аурулар. Тұқым қуалайтын аурулар гендік, хромосомалық және геногеномдық мутациялардың әсерінен генетикалық материалдың өзгеруіне байланысты қалыптасады. Генетикалық жіктеу бойынша тұқым қуалайтын аурулар: • моногендік; • хромосомалық; • мультифакторлық (полигендік) болып бөлінеді. Моногенді аурулар генетикалық ақпарат жазылған құрылымдық гендердің мутацияға ұшырауынан туындайды. Бұл аурулардың ұрпақтарға берілуі Г. Мендельдің тұқым қуалау заңдылықтарына сәйкес жүретіндіктен мендельденуші тұқым қуалайтын ауру деп аталады. Моногенді түрі аутосом. -доминантты (арахнодактилия, брахидактилия, полидактилия, т. б. дерттер), аутосом. -рецессивті (екі, кейде үш немере ағайынды некелескен адамдар арасында жиі кездеседі; агаммаглобулинемия, алкаптонурия, т. б. дерттер) және жыныстық Х- және У-хромосомалармен тіркескен (генге байланысты еркек ауырады, ал ауруды әйел адам тасымалдайды; гемофилия, т. б. дерттер) тұқым қуалайтын аурулар болып бөлінеді. Хромосомалық аурулар геномдық (хромосомалар санының өзгеруі) және хромосомалық (хромосомалар құрылысының өзгеруі) мутацияларға байланысты қалыптасады. Жиі кездесетін хромосома ауруларының қатарына трисомиялар жатады. Бұл кезде хромосома жұптарының бірінде қосымша 3-хросома пайда болады. Мысалы, Даун ауруында аутосом. 21-жұп бойынша трисомия болса, Патау синдромында 13-жұпта, Эдварс синдромында 18-жұбында болады. Гаметогенезде мейоздық бөлінудің бұзылуына байланысты әйелдерде жыныстық Х - хромосомалардың біреуі болмаса, Шерешевский-Тернер синдромы, керісінше бір хромосом артық болса - трипло-Х (ер адамдарда Клайнфельтер) синдромының қалыптасуына әкеледі. Жасы 35-тен асқан әйелдердің бала көтеруінде нәрестелердің хромосом. аурумен туу қауіптілігі жоғары болады. Мультифакторлық аурулар бірнеше геннің мутацияға ұшырауы мен өзара әрекеттесу нәтижесінде, ауруға бейімделуі артқан кезде және қоршаған орта факторларының әсеріне байланысты туындайды. Мұндай ауруларға • подагра; • қант диабеті; • гипертония; • асқазан және ішектің ойық жарасы; • атеросклероз; • жүректің ишемия ауруы, т. б. жатады. Тұқым қуалайтын аурулардың бұл түрінің пайда болу себебі әлі толықтай анықталған жоқ. Тұқым қуалайтын ауруларды клиникалық жіктеу патологиялық өзгерістерге ұшыраған органдар мен жүйелер бойынша жүргізіледі. Мысалы, жүйке және эндокриндік жүйенің, қан айналым жүйесінің, бауырдың, бүйректің, терінің, т. б. органдардың тұқым қуалайтын аурулары деп жіктеледі. Республикада тұқым қуалайтын ауруларды анықтау, емдеу жұмыстарымен неврология, терапия, хирургия клиникалар мен ауруханалар айналысады.

НЕгізгі бөлім
Гендік аурулар - генетикалық ақпарат жазылған құрылымдық гендердің мутацияға ұшырауынан туындайды. Бұл аурулардың ұрпақтарға берілуі Г. Мендельдің тұқым қуалау заңдылықтарына сәйкес жүретіндіктен мендельденуші тұқым қуалайтын ауру деп аталады. Олар 3 түрге жіктеледі:
1. Энзимопатия. Энзимопатияларға негізінде не фермент белсенділігінің өзгерісі, не оның синтезі қарқынының төмендеуі жатады. Гетерозиготаларда қалыпты аллелдермен компенсация есебінен көріне бермейді.
2. Гемоглобинопатиялар - гемоглобиннің пептидті тізбектерінің біріншілік ақауымен жəне осы бұзылысқа байланысты оның қасиеті мен қызметінің бұзылысынан болатын тұқым қуалайтын аурулар тобы. Мысалдар: метгемоглобинемиялар, эритроцитоздар, орақтəрізді-жасушалық анемия, талассемия.
3. Коллагенді аурулар. Коллаген биосинтезі мен ыдырауының генетикалық ақаулары. Бұл топқа Эллерс-Данлос ауруы, Марфан ауруы жатады.

Гемоглобинопатия
Гемоглобинопатия - гемоглобин ақуызының құрылымындағы тұқым қуалайтын немесе туа біткен өзгеріс немесе бұзылыс, әдетте оның оттегін тасымалдау қызметінде немесе эритроциттердің құрылымы мен қызметінде клиникалық немесе зертханалық түрде байқалатын өзгерістерге әкеледі.
Ең жиі кездесетін және белгілі гемоглобинопатияларға орақ жасушалы анемия, бета-таласемия және ұрықтың гемоглобинінің тұрақтылығы жатады.
Гемоглобинопатиялар сапалық және сандық болып бөлінеді.
Сапалылары полипептидтік тізбектердегі амин қышқылдарының орын ауыстыруына байланысты. β-тізбегіндегі глютамин 6 амин қышқылын валинмен алмастыру орақ жасушалы анемияның дамуының негізінде жатқан қалыптан тыс гемоглобин S түзілуіне әкеледі.
Гемин қалтасының аймағында (гемді бекіту орны) аминқышқылдарын алмастыру, ол метгемоглобин түріндегі молекуланы тұрақтандырады және эритроцитоз мен цианоздың дамуын тудырады;
Гемоглобин молекуласының тұрақсыздануын тудыратын (тұрақсыз Hb) альфа немесе бета тізбегіндегі аминқышқылдарының орын ауыстыруы немесе реттілігін өзгерту. Қалыпты тізбектерден тұратын атипті тетрамерлердің түзілуі (Hb-betta4, Hb-gamma4) .
300-ден астам нормадан тыс гемоглобиндер бар, бірақ барлығында ауытқулар пайда болмайды.
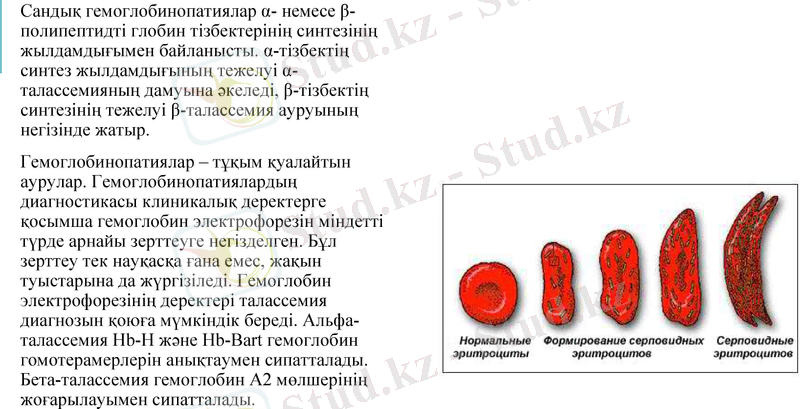
Сандық гемоглобинопатиялар α- немесе β-полипептидті глобин тізбектерінің синтезінің жылдамдығымен байланысты. α-тізбектің синтез жылдамдығының тежелуі α-талассемияның дамуына әкеледі, β-тізбектің синтезінің тежелуі β-талассемия ауруының негізінде жатыр.
Гемоглобинопатиялар - тұқым қуалайтын аурулар. диагностикасы клиникалық деректерге қосымша гемоглобин электрофорезін міндетті түрде арнайы зерттеуге негізделген. Бұл зерттеу тек науқасқа ғана емес, жақын туыстарына да жүргізіледі. Гемоглобин электрофорезінің деректері талассемия диагнозын қоюға мүмкіндік береді. Альфа-талассемия Hb-H және Hb-Bart гемоглобин гомотерамерлерін анықтаумен сипатталады. Бета-талассемия гемоглобин Α2 мөлшерінің жоғарылауымен сипатталады.

Гемоглобиннің қалыптан тыс пайда болуының осы механизмдеріне сүйене отырып, сапалы немесе құрылымдық келесі клиникалық жіктелуі ұсынылады:
1. Орақ тәрізді жасушалық ауру: SS-гемоглобинопатия және оның нұсқалары (S-талассемия, СК, SD, SE және т. б. .
2. Гомозиготалы гемоглопатиялар. (CC, DD, EE) қатерсіз ағымымен сипатталады
3. Цианозды және жиі эритроцитозды тудыратын гемоглобинопатиялар
4. Гемоглобиндердің болуына байланысты емес гемоглобинопатиялар және эритроциттерде Гейнц денешіктері бар сфероцитарлы емес гемолитикалық анемиямен сипатталады
5. Біріктіру және бета тізбектерінен туындаған лепорлы гемоглобинопатия және клиникалық тұрғыдан бета талассемияға ұқсайды
6. Констант-көктемгі гемоглобинопатия, көбінесе альфа тізбегі мен терминальді реаксацияға байланысты альфа тізбегінің ұзаруымен сипатталады және бастапқы орақ жасушалы анемия және талассемия көріністерінің ауырлығымен ерекшеленеді

Талассемия
Талассемия - қандағы оттегін тасымалдайтын эритроциттердегі ақуыз - гемоглобин түзілуінің бұзылуымен сипатталатын сирек кездесетін тұқым қуалайтын қан ауруы. Талассемияның негізгі көрінісі гемолиз болып табылады - қызыл қан жасушаларының жедел өлімі. Ауру тұқым қуалайтын гемолитикалық анемиялар тобына жатады.
Альфа талассемия. HBA1 және HBA2 гендерінің мутацияларымен байланысты. α-тізбектерді кодтайтын тек 4 локус бар. Локустардың бірінде мутацияның болуы минималды клиникалық көріністерге әкеледі. Екі локустағы бұзушылықтар анемияның жеңіл түрімен көрінеді. Үш локустағы мутация кезінде α-глобин өндірісінің айтарлықтай төмендеуі орын алады. Сонымен қатар артық β-глобин тізбектері тетрамерлерді құрайды - гемоглобин H. Бұл нысан гемоглобинопатия H деп те аталады. Аурудың сипаты жеңілден ауыр гипохромды микроцитарлық анемияға дейін өзгеруі мүмкін. Альфа-глобиннің барлық төрт аллельінде мутацияның болуы өмірмен үйлеспейді. Мұндай патологиясы бар бала жатырда немесе туғаннан кейін көп ұзамай өледі.
Бета талассемия. Бета талассемияның екі нұсқасы бар - негізгі талассемия және кіші талассемия (кіші), олардың ішінде негізгі талассемия аурудың ең ауыр түрі болып табылады. Бета-глобин генінің екі аллельінде де мутация болған кезде пайда болады. Бета тізбектерінің өндірісі болмаған немесе күрт төмендеген кезде гемоглобин А гемоглобин F-ге ауыстырылады, ол әдетте ұрықта өндіріледі және босанғаннан кейін А гемоглобинімен ауыстырылады. Кіші Талассемия бета-глобин генінің аллельдерінің бірінде мутацияның болуымен байланысты. Әдетте, ол оңай жүреді және емдеуді қажет етпейді.
Көп жағдайда талассемия балалық шақта диагноз қойылады. Дегенмен, аурудың асимптоматикалық ағымының жағдайлары бар, бұл кезде диагноз ересек жаста жоспарлы қан анализінен кейін ғана қойылады. Аурудың кейбір түрлері іс жүзінде емдеуді қажет етпейді, басқалары донорлық қанды тұрақты құюды, кейбір жағдайларда көкбауырды алып тастауды талап етеді.

Клиникалық көрінісі
Талассемия белгілері өте ауыр және айқын. Жоғарыда айтылғандай, бұл ауру анемиямен бірге жүреді. Егер анемия ерте жаста дамыған болса, онда мұндай балада психика дамымаған, сондай-ақ физикалық дамудың бұзылуы бар. Сондай-ақ, қызыл қан жасушаларының ерте бұзылуы сүйек кемігінің гиперплазиясына әкелетінін атап өткен жөн. Мұндай өзгерістер бас сүйегінің дамуының бұзылуына әкеледі. Бас сүйегінің төртбұрышты немесе биік болып байқалады, тістеу бұзылыстары, мұрын пішіні жалпақ бола алады. Талассемияда жанама билирубин синтезінің жоғарылауына байланысты сарғаю байқалады. Сондай-ақ, талассемиямен, әдетте, көкбауыр ұлғаяды. Қолданыстағы клиникалық көрініске байланысты, талассемиясы бар науқастар жұқпалы ауруларға өте сезімтал. Ауыр анемия жағдайында балалар өмірінің бірінші жылында жиі өледі. Ауыр емес формаларда ересек жасқа дейін аман қалуға болады.

Диагностикасы
Диагностика науқаста талассемияға күдіктенген кезде жүргізілетін зерттеулерді екі топқа бөлуге болады:
Гемолизді диагностикалау үшін қажет (эритроциттердің жедел өлімі)
Талассемияны диагностикалау және аурудың түрін анықтау үшін қажет.

Гемолизді диагностикалау үшін келесі зерттеулер жүргізіледі:
Жалпы қан анализі. Гемолиздің болуы қызыл қан жасушалары мен гемоглобин деңгейінің төмендеуін көрсетуі мүмкін. Талассемия сонымен қатар MCH (эритроциттегі гемоглобиннің орташа мөлшері) - гипохромия, MCV (эритроциттердің орташа көлемі) - микроцитоз мәндерінің төмендеуімен сипатталады. MCH және MCV мәндерінің төмендеуі темір тапшылығы анемиясында да байқалуы мүмкін.
Қандағы ретикулоциттер. Ретикулоциттер деңгейінің жоғарылауы науқаста гемолиздің болуын көрсетуі мүмкін.
Билирубин. Бос (жанама) және жалпы билирубин деңгейінің жоғарылауы науқаста эритроциттердің жедел өлімінің болуын болжауға мүмкіндік береді.

Талассемияның болуын диагностикалау және оның түрін анықтау үшін келесі зерттеулер жүргізіледі:
Қан сарысуындағы гаптоглобин. Гаптоглобин - қызыл қан жасушалары өлгеннен кейін қанға түсетін гемоглобинді бұзатын ақуыз. Гаптоглобин деңгейінің төмендеуі талассемияның болуын көрсетуі мүмкін.
Қан жағындысының микроскопиясы. Бұл микроскоптың астындағы науқастың қанын зерттеу, ол төмендеген қызыл қан жасушаларының, сондай-ақ талассемияға тән болатын пішіні өзгерген эритроциттердің болуын анықтауға мүмкіндік береді.
Гемоглобин электрофорезі. Бұл зерттеу талассемияны диагностикалаудың негізгі әдісі болып табылады. Гемоглобин электрофорезі қандағы гемоглобиннің әртүрлі түрлерінің (HbA, HbA 2, HbF), оның ішінде қалыпты емес - HbH болуын анықтауға мүмкіндік береді.
Генетикалық кеңес беру. Талассемия тұқым қуалайтын ауру болғандықтан, ұрықтың даму сатысында Барт гемоглобинінің болуын анықтауға дейін ұрпақта талассемияның даму мүмкіндігін анықтау үшін бірқатар генетикалық зерттеулер мен сынақтар бар.

Емі
Қызыл қан клеткаларын құю. Талассемияның ауыр түрлерінде эритроциттік қан препараттарын құю қажеттілігі өмірдің алғашқы айларынан бастап туындайды және әртүрлі дәрежеде болса да, өмір бойы сақталады - трансфузиялық тәуелділік деп аталатын дамиды. Бұл пациенттердің қанындағы гемоглобиннің төмендей беруін тоқтатпайтынын және оның алдын алу үшін жоғарылатудың басқа нақты жолдары жоқ екенін білдіреді, тек мұндай құюлардан басқа. Науқастың қанындағы гемоглобин мөлшері төмен деңгейге дейін төмендемегені жөн, қанның мөлшерінің қанағаттанарлық г/л деңгейінде қайта құйған жөн.
Десферал
Емдеудің маңызды бөлігі «десферал» препаратымен жүзеге асырылатын «хелаттар» тобынан («хелат терапиясы» деп аталатын) препараттардың көмегімен денеден артық темірді жою болып табылады. Қазіргі уақытта емдеу көптеген сағаттар бойы тері астына инъекциялар арқылы қабылданады, ең қолайлысы арнайы құрылғыларды пайдалану болып табылады. Помпаға бекітілген шприцтен Десферал бірнеше сағат бойы пациентке тері астына біртіндеп енгізіледі. Ең дұрысы, ауыр талассемиямен ауыратын науқастар өмір бойы аптасына 5 күн десфералды қабылдауы керек, бірақ нақты өмірде бұған жету әлі де қиын.

Көкбауырды алып тастау (спленэктомия)
Кейбір науқастарда көкбауырдың өте үлкен мөлшерінің өзі гемоглобиннің күйіне және қан жүйесінің басқа көрсеткіштеріне теріс әсер ете бастайды. Мұндай жағдайларда ол хирургиялық жолмен жойылады. Бұл операция талассемияның өзін емдей алмайды, бірақ ол оның көріністерін жеңілдетуі мүмкін (бірақ олай болмауыда мүмкін) . Спленэктомия көкбауыр өте үлкен болғанда, сонымен қатар оның қанның басқа көрсеткіштеріне патологиялық әсерінің айқын белгілері болған кезде ғана жасалады («гиперспленизм» деп аталады) . Операция 5 жасқа толғанға дейін ұсынылмайды, 8-10 жас оңтайлы болып саналады.

Сүйек кемігін трансплантациялау
Қазіргі уақытта сүйек кемігін трансплантациялау арқылы талассемияны емдеу кең таралған. Бұл талассемияны түбегейлі емдеудің жалғыз әдісі. Талассемия анықталған кезде, сүйек кемігінің донорын табу үшін пациенттер мен олардың отбасыларының «HLA типті» болуы (яғни, күрделі биологиялық үйлесімділік сынағын өткені жөн) болғаны жөн. Дегенмен, қолайлы донорды табу әдетте қиын, ал үйлесімді туысы жоқ донорды табу процесі әлі де қымбат және көп уақытты қажет етеді. Сүйек кемігін трансплантациялаудың өзі өте қымбат. Жақын туыстары, тіпті егер олар HLA антигендерімен үйлесімді болса да, көбінесе талассемиямен ауырады. Сондықтан талассемиямен ауыратын салыстырмалы түрде аз пациенттер әлі де сүйек кемігін трансплантациялау арқылы емдеуге нақты үміткерлер болып табылады.

Орақ жасушалы анемия
Орақ жасушалы анемия -тұқым қуалайтын глобин синтезінің бұзылыстарына байланысты оттегі беретін глобин ерігіштігінің төмендеу салдарынан эритроциттер пішіні орақ тәріздес өзгеруімен жүретін дерт. Орақтәрізді эритроциттердің пайда болуы қан тұтқырлығының жоғарылауына, қан ағысының баяулауына және капиллиярда қан іркілуіне алып келеді. Қан іркілісі гипоксияны туындатып, эритроциттердің орақ тәрізді өзгеруінің тереңдеуіне себепкер болады. Гемоглобин S концентрациясы өте жоғары эритроциттерде оттегінің парциалды қысымы 60 мм с. б. төмендегенде олар орақ тәрізді өзгерсе, гемоглобин S концентрациясы 25-50 % болған жағдайда оттегінің парциалды қысымы 10 мм с. б. жеткенде ғана олар орақ тәрізді өзгереді екен.

Этиологиясы және патогенезі
- Іс жүргізу
- Автоматтандыру, Техника
- Алғашқы әскери дайындық
- Астрономия
- Ауыл шаруашылығы
- Банк ісі
- Бизнесті бағалау
- Биология
- Бухгалтерлік іс
- Валеология
- Ветеринария
- География
- Геология, Геофизика, Геодезия
- Дін
- Ет, сүт, шарап өнімдері
- Жалпы тарих
- Жер кадастрі, Жылжымайтын мүлік
- Журналистика
- Информатика
- Кеден ісі
- Маркетинг
- Математика, Геометрия
- Медицина
- Мемлекеттік басқару
- Менеджмент
- Мұнай, Газ
- Мұрағат ісі
- Мәдениеттану
- ОБЖ (Основы безопасности жизнедеятельности)
- Педагогика
- Полиграфия
- Психология
- Салық
- Саясаттану
- Сақтандыру
- Сертификаттау, стандарттау
- Социология, Демография
- Спорт
- Статистика
- Тілтану, Филология
- Тарихи тұлғалар
- Тау-кен ісі
- Транспорт
- Туризм
- Физика
- Философия
- Халықаралық қатынастар
- Химия
- Экология, Қоршаған ортаны қорғау
- Экономика
- Экономикалық география
- Электротехника
- Қазақстан тарихы
- Қаржы
- Құрылыс
- Құқық, Криминалистика
- Әдебиет
- Өнер, музыка
- Өнеркәсіп, Өндіріс
Қазақ тілінде жазылған рефераттар, курстық жұмыстар, дипломдық жұмыстар бойынша біздің қор #1 болып табылады.

Ақпарат
Қосымша
Email: info@stud.kz