Адамның тұқым қуалайтын аурулары: моногенді және полигенді аурулар - тәжірибелік сабақтарға әдістемелік нұсқау

Ф - ОБ - 012/016
Қ. А. Ясауи атындағы Халықаралық қазақ - түрік университеті
№1 Медицина факультеті
Адам морфологиясы кафедрасы
Молекулалық биология және медициналық генетика пәнінен тәжірибелік сабақтарға арналған әдістемелік нұсқаулар
Тақырыбы: 26 - 27 АДАМНЫҢ ТҰҚЫМ ҚУАЛАЙТЫН АУРУЛАРЫ.
МОНОГЕНДІК ЖӘНЕ ПОЛИГЕНДІК АУРУЛАР
Құрастырған: фарм. ғ. к., доцент м. а. М. А. Бердібеков
а/ш докторы, доцент К. М. Лаханова
оқытушы: Б. Т. Тастемирова
Түркістан - 2011ж.
Кафедра мәжілісінде талқыланды, хаттама № ___ , “” 2011ж. Кафедра меңгерушісі, м. ғ. д., профессор С. Н. Жұмашов1. Тақырып: АДАМНЫҢ ТҰҚЫМ ҚУАЛАЙТЫН АУРУЛАРЫ.
МОНОГЕНДІК ЖӘНЕ ПОЛИГЕНДІК АУРУЛАР
2. Тақырыптың өзектілігі: Адамзаттың ауруы мен өлімінде тұқым қуалаушылықпен анықталатын адамның патологиялық жағдайы маңызды роль алады, әсіресе дамыған елдерде. Генетикалық өзгерістердің жаңа туылған балалар арасындағы жиілігі 3-5 %, хромосомалық аурулар 0, 5%, моногендік аурулар - 1%, мультифакторлық аурулар-2-3% құрайды. Жастың ұлғаюына байланысты олардың жиілігі өсуде. Соңғы жылдары дүние жүзіндегі балалар аурулары мен өлімі құрылымында генетикалық патологияның өсу тенденциясы байқалады. Әсіресе, перинатальдық және нәрестелердің өлімінің 20-30 % жағдайлары генетикалық өзгерістен болады. Ауруханаларға жатқызылған ауру балалардың 30-40% дейінгі себептері тұқым қуалайтын патологиялар. Жүктілік үзілуінде (өздігінен болған түсіктерде) алғашқы бедеулікте, жүйке, эндокриндік, жүрек-тамыр, жыныс және т. б. жүйелердің патологиясында генетикалық фактор маңызды роль атқарады.
Адамның хромосомалық ауруларының жалпы саны 100-ге жетеді. Ал моногендік аурулар - 1 нозологиядан асады. Бұл мәліметтер қазіргі кездегі дәрігерлерге адамның тұқым қуалайтын патологиясының пайда болу себептерін, даму механизмдерін (патогенезін), диагностикалық әдістерін, емдеуін және профилактикасын зерттейтін медициналық (клиникалык) генетиканың негіздерін білудің маңыздылығын көрсетеді.
3. Сабақтың мақсаты: Студенттерді гендік аурулардың клиникалық белгілерімен, оларды диагностикалау әдістерімен және алдын алуымен таныстыру.
4. Оқытудың міндеттері:
5. Тақырыптың негізгі сұрақтары:
а) бастапқы білімдері бойынша:
1. Жасушаның тұқым қуалау аппараты.
2. ДНҚ молекуласының құрылысы.
3. Гендердің маңызы бойынша жіктелуі.
4. Өзгергіштік және оның формалары.
5. Мутациялық өзгергіштік және оның жіктелуі. Гендік мутациялар, олардың маңызы.
б) сабақтың тақырыбы бойынша:
1. Гендік аурулар және олардың жіктелуі.
2. Адамның гендік аурулары дамуының себептік механизмдері.
3. Адамның гендік ауруларының диагностикасы.
4. Гендік аурулардың алдын-алу және емдеу жолдары.
6. Білім берудің және оқытудың әдістері:
- Негізгі сұрақтар бойынша ауызша талдау.
- Типтік және ситуациялық есептерді шығару.
- Пікірталас.
7. Тәжірбиелік сабақтың өтілетін орны: Адам морфологиясы кафедрасының оқу бөлмелері: 202, 204, 205, 206.
Ақпараттық - дидактикалык блок:
Гендік мутациялар тудыратын аурулар гендік аурулар деп аталады.
Жеке гендердің мутацияларына байланысты болатын тұқым қуалайтын аурулардың саны 10 000-нан асады. Алайда, олардың аурушылдық пен елімнің жалпы санындағы үлесі аз, 1 пайызға жуык болады. Гендік аурулардың жартысынан көбін моногенді аурулар құрайды. Олардың пайда болуы мутантты геннің әсеріне байланысты және патогенезі бір геннің алғашқы өніміне (ақуыздың, ферменттің болмауына, немесе олардың аномальды құрылымына, немесе аз синтезделуіне, немесе артық синтезделуіне) байланысты. Гендік аурулардың пайда болуы механизмінің жалпы заңдылығын нұсқа түрінде былай көрсетуге болады: мутантты ген -> патологиялық алғашқы өнім (ақуыз, фермент) -> биохимиялық реакция -* мүше ->ағза.
Гендік аурулардың кеңінен таралғаны мультигендік (мультифакторлық) аурулар, олардың дамуына бірнеше гендер үлес қосады, бірақ гендердің патологиялык әсері тек сыртқы ортаның белгілі бір қолайсыз жағдайларында іске асырылады.
Мутацияға ұшыраған локустардың (гендердің) санына байланысты гендік ауруларды моногендік (монофакторлық) және полигендік (мультифакторлық) деп жіктейді.
Моногендік ауруларда генетикалық эффект хромосоманың бір локусындағы мутациямен байланысты. Олардың негізінде жеке гендердің мутациясы, соның ішінде ДНҚ молекуласындағы нуклеотидтердің ретінің немесе құрамының өзгеруіне, генетикалық ақпараттың трансляциясының бұзылуына алып келетін экзондардың және т. б. дефектісі жатады.
Моногендік аурулар мендельдік заңдылықтар бойынша аутосомды доминантты, аутосомды рецессивті, X- тіркескен доминантты, X-тіркескен рецессивті, У - тіркесіп тұқым қуалайды.
Аурулардың тұқым қуалауы жыныспен (Х-немесе У-хромосомамен) тіркесуімен қатар, жыныспен шектелуі де мүмкін. Мысалы, бастың "таз", шаштың болмауы жынысқа байланысты еркектерде байқалады, әйелдерде болмайды.
Моногендік аурулар жаңа туылған 1000 нәрестеге шакқанда 42-65 балада кездеседі, бұл 4, 2-6, 5% болады, ал 5 жасқа дейінгі балалардың жалпы өлім себептерінің арасында моногенді аурулар 8-10% құрайды.
Моногендік аурулардың диагностикасында қолданылатын әдістер: генеалогиялық, биохимиялык, микробиологиялық, молекулярлы-генетикалық және пренатальдық (инвазивтік және инвазивтік емес)
Моногендік аурулардың жіктелу приициптері
Тұқым қуалау типіне байланысты:
1. Аутосомды-доминантты тип: ахондроплазия, миотониялық дистрофия, остеогенездің жетіспеушілігі, ретинобластома, Морфан синдромы, Гентингтон хореясы және т. б.
2. Аутосомды-рецессивті тип: алкаптонурия, альбинизм, атаксия, телеангиоэктазия, Тея-Сакс ауруы, галактоземия, муковисцидоз.
3. Х-тіркескен, доминантты тип: гипофосфатемия, Д витаминге төзімді мешел ауруы.
4. Х-тіркескен, рецессивті тип: А- және В-гемофилия, Дюшен-Беккер миодистрофиясы, Леш-Найян синдромы, глюкоза-6-фосфат-дегидрогеназа және т. б.
Мүшелік және жүйелік типі бойынша:
1. Жүйке жүйесінің аурулары;
2. Жүрек-қан тамырлар жүйесі және т. б. Этиологиясы бойынша:
1. Бастапқы дефектісі анықталған аурулар;
2. Бастапқы дефектісі анықталмаған аурулар (барлық моногендік аурулардың 90% жуығы) .
Зат алмасу түрінің бұзылуы бойынша:
Олар биохимиялық реакцияның өзгеруімен байқалады және хромосомаларды микроскопиялық зерттегенде көрінбейді.
Көмірсу алмасуының бұзылуына мысалға галактоземия, фруктозурия; май алмасуының бұзылуының нәтижесінде Тея-Сакс синдромы, ксантоматоз; аминқышқылы алмасуының бұзылуынан феиилкетонурия, альбинизм; минералдар алмасуының бұзылуынан Пильсон-Коновалов ауруы және т. б. жатады.
Зат алмасуының бұзылуынан басқа гендік мутациялар белоктардың құрылымын өзгертеді, мысалы гемоглобинопатиялар: орақ тәрізді жасушалық анемия, а- және β-талассемия; сонымен қатар түрлі морфологиялық өзгерістерге: ахондроплазияға, брахидактилияға, полидактилияға, анэнцефалияға жәнет. б. әкеледі.
70 жылдары митохондриялық және пероксисомалық аурулар анықтадды, олар тезаурисмоздармен бірге жасуша органеллаларындағы зат алмасу патологиясына жатады.
Полигендік (мультифакторлық) аурулар - хромосоманың бірнеше локусындағы мутациясының бірлесіп әсер етуіне байланысты болады. Бұл жағдайда генетикалық эффект бейімділікті тудырады, ал аурудың пайда болуы генетикалық және орталық факторлардың күрделі әсерлесуіне байланысты. Оларға псориаз, шизофрения, қант диабеті және т. б. жатады.
Мультифакторлық белгілерге ұдайы өзгерумен сипатталатын (бой ұзындығы, дене салмағы және т. б. ) белгілер және туа пайда болатын ақаулыкггар, сонымен қатар созылмалы, кеңінен таралған инфекциялық емес аурулар жатады. Белгілердің көрінуіндегі генетикалық және орта факторларының әсер ету деңгейін анықтау үшін егіздерді салыстыру әдісі қолданылады (2 кесте) .
2 - кесте
Тұқым қуалаушылықтың әртүрлі типтеріндегі егіздердің конкорданттылығы ( Ф. Фогель, А. Мотульски, 1989 ж) .
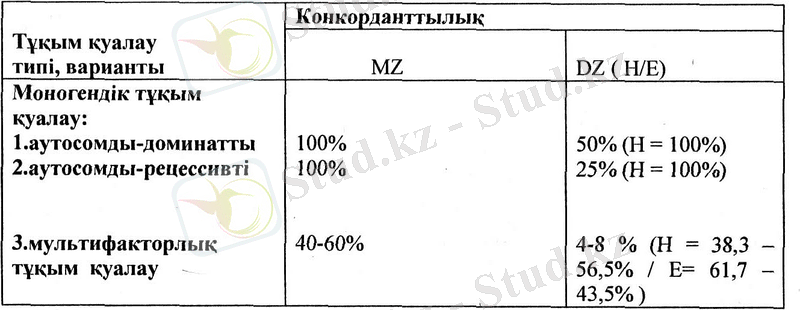
Кестеден моногендік аурулардың пайда болуында генетикалық факторлар әсерінің маңыздылығы көрінеді; Н мәні 100% тең, ал полигендік ауруларда Н = 30 - 60%. Полигендік (мультифакторлық аурулардың пайда болуы орта факторларының әсеріне байланысты (Е= =62 - 44%, оларда тек әртүрлі деңгейдегі бейімділік тұқым қуалайды (Н = 30 - 60%)
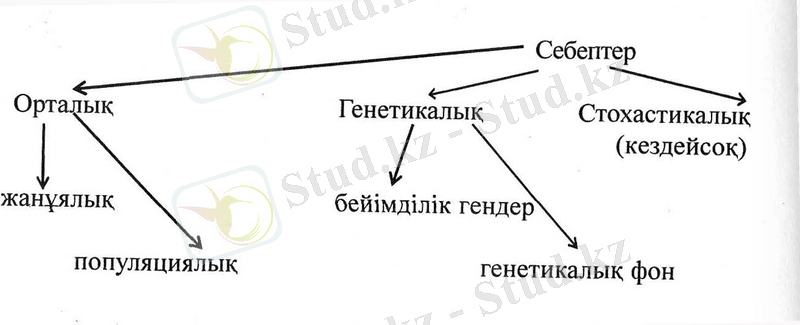
МУЛЬТИФАКТОРЛЫҚ АУРУЛАРДЫҢ СЕБЕПТЕРІ
8. Бақылау:
Студенттердің өзіндік жұмыстарының орындалуын бақылау
Тақырыптық негізгі сұрақтары бойынша сұрау
Тесттік бақылау
1-тапсырма. "Моногенді аурулар" дидактикалық карталарымен жұмыс істеу. Гендік аурулардың клиникалық белгілерімен танысып, анықтау әдістерін тауып және диагноз қойыңыз.
№1
. . . туа біткен дефектісіне
байланысты ауру. айналу процесінің бұзылуы қанда фенилаланиннің жиналуына әкеледі, соған байланысты түрлі патологиялық құбылыстар пайда болады. Әр елдегі оның таралу жиілігі 1:2600 (Турцияда) - 1:30 000 (Швецияда) тең, тек Жапония мен Финляндияда өте сирек кездеседі.
. . . ауруы жаңа туылған нәрестелерде байқалмайды, олардың сыртқы көрінісі қалыпты, бірақ өмірінің алғашқы аптасында патологияның неврологиялық белгілері байқалады: жоғарғы тітіркенгіштік, сіңір рефлекстерінің күшеюі, бұлшық ет гипертонусы, дірілдеу, тырыспа, диспепсия. Кейінірек, өмірінің 4-5 айларында ақыл-естің жетілуі нашарлайды, микроцефалия, тері жамылғысы, шашы, көздің нұрлы қабығы бозарады.
№2
. . . тирозиназа ферменті катализдейтін тирозиннен 3-4 диоксифенилаланин арқылы меланин түзілуінің мутациялық блокадасынан пайда болады. Оның негізгі белгілері: тері, шаш, көздің нұрлы қабығы жасушаларында меланиннің болмауы және УК-сәулеге сезімталдықтың жоғарылауы. Бұл патологияның түрлі регион популяциясындағы жалпы жиілігі-1:5000-нан 1:25000 дейін.
. . . алтыға жуық түрлі генетикалық формасы ажыратылады, кейбіреулері өте сирек кездеседі: олардың генетикалық гетерогендігіне пигментациясы бойынша қалыпты балалары бар екі альбиностын арасындағы некелер көрсетеді. Кейбір формаларының клиникалық ерекшеліктері (мысалы, жас мелшеріне байланысты белгілі пигменттелу ерекшелігі, қансыраудың жоғарылауы) болады.
№3
. . . сирек кездеседі (3-1: ) . Клиникалык белгілері
40 жастан және одан кейін басталады: аяқ-қол буындарындағы, омыртқа жотасындағы және басқа да дәнекер ұлпаға бай дене беліктеріндегі патологиялық өзгерістермен сипатталады. Патологияның негізінде охра пигментін түзетін гомогентизин қышқылының дәнекер ұлпаға жиналуы жатады. Қышқылдың көп мөлшері зәрмен сыртқа шығарылады, ауада тұрған зәр қараяды.
. . . гомогентизин қышқылын малеинацетосірке қышқылына айналуын катализдейтін окзидаза ферментінің генетикалық дефектісіне байланысты.
. . . ағылшын дәрігері А. Гаррод (1902 ж) зер1-еп, туа пайда болатын тұқым қуалайтын зат алмасу ауруы екенін анықтады. Гомогентизин қышқылының ыдырауына қатысатын оксидаза ферментінің синтезделуін бақылайтын структуралық геннің полиаллелизміне байланысты бұл ауруға да генетикалық гетерогенділік тән.
№4
. . . көмірсулар алмасуы аномалияларының тобына
жатады, оларға орталық нерв жүйесінің, бұлшық ет жүйесінің жарақатгануы, бауыр қызметінің бұзылуы, эритроцитгердің аномалиясы, гипогликемиялық жағдайлар тән . . . кезінде алғашқы биохимиялық дефекттің негізінде галактоза-1-фосфат-уридилтрансферазаның жетіспеушілігі (дефициті) жатады, осыған байланысты ағзаның ұлпаларында галактоза-1-фосафаттың аса көп мөлшері және лактозаның толық емес ыдырауының басқа заттары жиналады, олар клиникалық белгілерін тудырады.
Нәрестелердегі аурудың жиілігі 1:3, кейбір авторлардың мәліметі бойынша 1:8000 дан 1:187000 дейін кездеседі.
Аурудың алғашқы симптомдары туылысымен, балалардың сүт емуінен басталады: құсады, іші өтеді, дене салмағы азаяды, сарғаяды, тырыспа және гипогликемия пайда болады. Біртіндеп катарактаның, бауыр циррозының белгілері байқалады және ақыл-естің дамуы нашарлайды. Зілді формаларында өліммен аяқталады.
№5
Сфинголипидтердің жиналуынан болатын аурулардың клиникалық белгілеріне мидағы, сүйектегі және паренхиматоздық мүшелердегі (бауыр, көк бауыр, бүйрек), терідегі, көздің торлы қабатындағы өзгерістер нәтижесінде ақыл-естің және тірек қимылдың үстемеленіп бұзылуы тән. Бұл ауру сирек кездеседі, 3 туылған нәрестелердің
біреуінде кездеседі, ал арасында . . . ауруы еврей-ашкенази
(1:3600 нәрестелерде) өте жиі кездеседі. Барлық сфинголипидоздар аутосомды рецессивті типте және Х-хромосомамен тіркесіп тұқым қуалайды. Еврей-ашкенази популяцияларында аурудың алдын-алу мақсатында гетерозиготаларды анықтайды.
Сфинголипидтер жасушалық мембраналардың, әсіресе, нерв талшықтарының миелин қабығының маңызды құрылымдық компоненті, сондықтан олардың ағзада ұдайы жүретін жаңаруы, олардың жасуша лизосомаларындағы ыдырауы көптеген тіршілікке маңызды мүшелердің, мидың (сұр және ақ затының) жарақаттануының патологиялық белгілерін қалыптастырады. Сфинголипидтердің алмасу дефектісі нақты бір сфинголипидтердің типіне тән гексозаминидаза А ферментінің белгілі түрінің жетіспеуіне байланысты.
№6
. . . синдромы сирек кездеседі (1:3 нэрестеге) және X хромосомамен тіркескен рецессивті типті тұқым қуалайды. Бастапқы дефекті анықталған: ДНҚ синтездеуге қажет фермент гипоксантин-фосфорибозил-трансферазаның (ГФРТ) жетіспеушілігі, ол бос пуриндік негіздерді (гуанин және гипоксантинді) нуклеотидтерге айналуын катализдейді.
Ауру емізулі кезеңінен басталады; бұлшық еттің гипертонусы және олигофрения байқалады, рефлекторлық тітіркенгіштік жоғарылайды, бала өзін-өзі жарақатгайды. Ересек адамдарда бұл ауру өзгерген (типтік емес) формаларында: неврологиялық және подаграның белгілері түрінде кездеседі. Ферменттің жетіспеушілігінен негіздердің ақырғы ыдырау өнімінен зәр қышқылы пайда болады. Осы қышқылдың және оның тұздарының зәрмен көп бөлінуіне қарамай, олардың қандағы мөлшері жоғарылайды, біртіндеп ураттардың түзілуіне және бүйректе тастардың пайда болуына әкеледі.
№7
. . . ауруында мыстың алмасуы бүзылады, кездесу жиілігі
1:87000 (Жапониядағы туыс және туыс емес некелер арасында), мыстың қосындылары зат алмасу процессінде үлкен роль атқарады. Мыс тотығу реакцияларына қатысатын көптеген ферменттердің құрамына кіреді, олар митохондриялардың мембраналар құрамына енген және тотығу реакцияларының маңызды этаптарына қатысады.
Бұл ферменттің жетіспеуі жасушалық және ағзалық деңгейдегі ауыр өзгерістерге әкеледі. Белгілі гендік мутацияда мысты байланыстыратын белок церуллоплазминнің синтезделуі тоқтайды. Осыған байланысты мыстың қандағы мөлшері артады, бауыр, бүйрек және ми ұлпасында жиналып, . . . ауруын қоздырады.
Клиникалық сипаты: бауырда цирроз пайда болады, бүйрек және ми жасушалары өледі. Бүйректің қызметінің нашарлауы аминқышқылдарының, глюкозаның зэр кышқылының және фосфаттардың тасмалдануын бұзады.
№8
. . . бұл гемоглобинопатияда гемоглобин тізбектерінің (а
немесе (3) біреуінің синтезі бүзылады. р-тізбектер синтезінің бұзылуы жиірек байқалады және олардың орнына а тізбектерінің синтезделуі сақталады (НВС немесе НВА 2 ) . Р-тізбектердің жеткіліксіз аз түзілуі а-тізбектің артық (көбірек) түзілуіне әкеледі. Бұл жағдайда тұрақсыз (стабильді емес) гемоглобин түзіледі де, преципитацияланады және эритроциттерде денешіктер түрінде кездеседі. Мұндай эритроциттер тез жойылады және гемолизденеді.
Гомозиготаларда анемияның зілді формасы-үлкен . . . немесе
... жалғасыСіз бұл жұмысты біздің қосымшамыз арқылы толығымен тегін көре аласыз.
- Іс жүргізу
- Автоматтандыру, Техника
- Алғашқы әскери дайындық
- Астрономия
- Ауыл шаруашылығы
- Банк ісі
- Бизнесті бағалау
- Биология
- Бухгалтерлік іс
- Валеология
- Ветеринария
- География
- Геология, Геофизика, Геодезия
- Дін
- Ет, сүт, шарап өнімдері
- Жалпы тарих
- Жер кадастрі, Жылжымайтын мүлік
- Журналистика
- Информатика
- Кеден ісі
- Маркетинг
- Математика, Геометрия
- Медицина
- Мемлекеттік басқару
- Менеджмент
- Мұнай, Газ
- Мұрағат ісі
- Мәдениеттану
- ОБЖ (Основы безопасности жизнедеятельности)
- Педагогика
- Полиграфия
- Психология
- Салық
- Саясаттану
- Сақтандыру
- Сертификаттау, стандарттау
- Социология, Демография
- Спорт
- Статистика
- Тілтану, Филология
- Тарихи тұлғалар
- Тау-кен ісі
- Транспорт
- Туризм
- Физика
- Философия
- Халықаралық қатынастар
- Химия
- Экология, Қоршаған ортаны қорғау
- Экономика
- Экономикалық география
- Электротехника
- Қазақстан тарихы
- Қаржы
- Құрылыс
- Құқық, Криминалистика
- Әдебиет
- Өнер, музыка
- Өнеркәсіп, Өндіріс
Қазақ тілінде жазылған рефераттар, курстық жұмыстар, дипломдық жұмыстар бойынша біздің қор #1 болып табылады.

Ақпарат
Қосымша
Email: info@stud.kz