Алматы қаласында 2014 жылы туған нәрестелердің қанындағы ТТГ деңгейін иммунофлуориметриялық өлшеу және туа біткен гипотиреоздың кездесу жиілігі: ретроспективті талдау

БЕЛГІЛЕУЛЕР МЕН ҚЫСҚАРТУЛАР
ГП- Гипотиреоз
ДЙТ- Дийодтирозин
МЙТ- Монойодтирозин
НЙС- Натрий-йодты симпортер
Т3- Трийодтиронин
Т4- Тироксин
ТПО- Тиреоидты пероксидаза
ТТГ- Тиреотропты гормон
ФКУ - фенилкетонурия
КІРІСПЕ
Медициналық генетиканың дамуы молекулалық генетиканың ашқан ғылыми жаңалықтарымен тікелей байланысты. Ал молекулалық генетика - тұқымқуалайтын ауруларды молекулалық деңгейде зерттеп, оның молекулалық негізі мен механизмдерін анықтайды. Медициналық генетиканың негізгі мәселелерінің бірі - тұқымқуалайтын аурулардың молекулалық, биохимиялық механизмдерін анықтай отырып, оның емдеу жолдарын іздестіру. Негізінен қазіргі таңда тұқым қуалайтын көптеген аурулардың биохимиялық механизмдері анықталған. Әртүрлі себептердің әсерінен туындайтын тұқымқуалайтын ауруларды медициналық генетика саласы зерттейді [1, 2] .
Қазіргі таңда дүние жүзі халқын алаңдатып отырған мәселелердің бірі - йод жетіспеушілігі. Зерттеулерге сүйенсек, жер шарында бір миллиардтан астам адам табиғи йод жеткіліксіз аудандарда өмір сүреді. Осындай йод жеткіліксіздігі кездесетін мемлекеттердің қатарында Қазақстан да бар. Жағымсыз экологиялық жағдайлар йод жеткіліксіздігін туындата отырып, ол өз кезегінде тұрғындарды көптеген ауруларға ұшыратады, яғни адамдарда нәресте кезінен түрлі эндокриндік бұзылыстар туындайды. Осындай бұзылыстардың бірі - туа біткен гипотиреоз ауруы [3] .
Туа біткен гипотиреоз - балалардағы қалқанша безінің жиі кездесетін ауруы. Қазіргі таңда бұл ауру өзінің таралуы жағынан жас балалардың эндокринді ауруларының ішіндегі ең маңызды орын алатын ауруларының бірі. Туа біткен гипотиреоз - этиологиясы жағынан гетерогенді болып келетін, қалқанша безінің анатомиялық зақымдануынан, нәрестенің жатырішілік өмірінде гипоталамо-гипофизарлы жүйесінің бұзылуы немесе морфо-функциональды жағынан дұрыс жетілмеуінен туындайтын ауру. Ауру кезінде тиреоидты гормондардың жартылай немесе толық жетіспеушілігі байқалады. Ал тиреоидты гормондар организмнің қалыпты дамуына өте қажетті, себебі олардың жеткіліксіздігі баланың психикалық дамуын тежейді және жүйке жүйесінің түрлі бұзылыстарын туындатады. Бұл ауру қыз балаларда ұл балалармен салыстырғанда 2-2, 5 есе артық болып келеді. Әдебиеттердегі мәліметтерге қарағанда, туа біткен гипотиреоз ауруының кездесу жиілігі 1:3000 - 4000, кейде 1:5000 дейін ауытқиды [2, 4] . Ал отандық ғалымдардың зерттеулеріне қарағанда, Қазақстанда туа біткен гипотиреоз ауруының кездесу жиілігі - 1:3885 [5] .
Жұмыстың мақсаты: Алматы қаласында дүниеге келген нәрестелер қанындағы тиреотропты гормонның мөлшерін иммунофлуориметриялық зерттеу.
Жұмыстың міндеттері :
- Нәрестелер қанындағы тиреотропты гормон мөлшерін иммунофлуориметриялық зерттеу.
- 2014 жыл бойынша Алматы қаласындағы туа біткен гипотиреоз ауруының кездесу жиілігін анықтау.
- Туа біткен гипотиреоз ауруына ретроспективті талдау жасау.
Зерттеу жұмысының өзектілігі: бұл жұмыстың нәтижелері медицина, генетика және эндокринология саласына қосқан үлесі зор болып табылады. Себебі, нәрестелер қанындағы тиреотропты гормон мөлшерін анықтай отырып, туа біткен гипотиреоз ауруымен ауыратын нәрестелерді табу науқас нәрестенің болашақта арнайы ем шараларын қолданып сау болып дамуына мүмкіндік береді.
Ғылыми жаңалығы: Алматы қаласындағы туылған нәрестелердегі туа біткен гипотиреоз ауруының 2014 жыл бойынша кездесу жиілігі анықталды.
НЕГІЗГІ БӨЛІМ
1 ӘДЕБИЕТКЕ ШОЛУ
- Тұқым қуалайтын ауруларға сипаттама және олардың жіктелуі
Тұқым қуалайтын аурулар - тұқым қуалайтын ақпараттың өзгеруімен тікелей байланысты, ата-анадан ұрпаққа берілетін аурулар. Кез-келген тұқым қуалайтын аурудың өзіне тән клиникалық көрінісі болады. Тұқым қуалайтын аурулардың барлығына тән 5 жағдай:
- Ерте манифестация. Тұқым қуалайтын аурулардың 25%-ға жуығы нәрестеде туа пайда болса, 70%-ға жуығы жүре пайда болады және ауру 3 жастан асқаннан соң көріне бастайды, ал 5% жасөспірімдік кезеңде басталады.
- Созылмалы прогредиентті аралық. Прогредиентті дегеніміз науқастың ауру кезінде жалпы жағдайының нашарлауы мен жағымсыз белгілерінің пайда болуы. Созылмалы сипаты тұқым қуалайтын ауру кезінде мутантты геннің тұрақты қызметі анықталады.
- Зақымданудың көп болуы. 60% тұқымқуалайтын аурулар тек бір жүйені ғана зақымдап қоймай, қалған басқа да жүйелердің қызметтеріне өз зиянын тигізеді.
- Аурудың отбасылық сипатта болуы, бір аурудың бір отбасы мүшелерінде қайталанып кездесуі - ол аурудың тағы да туындайтынын анық көрсетеді. Бұл негізінен моногенді немесе мультифакторлы ауруға тән. Бірақ, хромосомалық синдром отбасыда тек бірақ адамда ғана кездеседі.
- Клиникалық полиморфизм. Аурудың клиникалық және зертханалық көрінісі көпжақты болса, онда ауруды бір-бірінен ажырату мүмкіндігі жоғары болады [3, 6] .
Барлық тұқым қуалайтын ауруларды 3 үлкен топқа ажыратамыз: моногенді, хромосомды, мультифакторлы. Моногенді ауру - ДНҚ молекуласы деңгейінде генетикалық ақпараттың зақымдануының нәтижесінде тек бір ғана ген бұзылады. Моногенді ауруларға көбінесе зат алмасу аурулары жатады. Олар: фенилкетонурия (ФКУ), галактоземия, муковицидоз, адреногенитиальды синдром, гликогеноздар, мукополисахариоздар және тағы басқалар. Моногенді аурулар Мендель заңына сәйкес тұқым қуалайды, ауру аутосомды-доминантты, аутосомды-рецессивті және Х-хромосоманың тіркесуі болып бөлінуі мүмкін. Хромосомды аурулар - геномдық және хромосомалық мутациялардың нәтижесінде пайда болады. Мультифакторлы аурулар - полигенді, қоршаған ортаның түрлі факторларының әсерінен бірнеше геннің мутацияға ұшырауынан мен өзара әрекеттесуі нәтижесінде туындайды. Мультифакториальды аурулардың жалпы белгілері мынадай:
1) тұрғындар арасында кездесу жиілігі жоғары;
2) клинкалық полиморфизм көрінген;
3) жақын туыстарда клиникалық көрінісінің ұқсас болуы;
4) жыныстық ерекшеліктер;
5) қарапайым Мендель үлгісінің тұқымқуалаушылық заңдылықтарына сәйкес келмеуі [6, 7] .
Көптеген ғалымдардың мәліметтері бойынша тұқымқуалайтын аурулардың жіктелуі 1- кестеде көрсетілген [6] .
Кесте 1.
Тұқымқуалайтын аурулардың жіктелуі [6] .
А. Сандық аномалия. Жыныс хромосомалары: Шерешевский - Тернер синдромы, Клайнфельтер синдромы, Х трисомия синдромы және т. б.
Аутосома:
Даун ауруы, Эдвардс және Патау синдромы және т. б.
Б. Хромосоманың құрылымдық аномалиясы: «мысық айғайы» синдромы және т. б.
А. Аутосомды-доминантты: Марфан синдромы, ахондроплазия, полидактилия және т. б.
Б. Аутосомды-рецессивті: ФКУ, галактоземия және т. б.
В. Х-тіркескен рецессивті: гемофилия, Дюшен миопатиясы және т. б.
Г. Х-тіркескен доминантты: Д- витамин - тұрақты рахит, тіс эмалінің қоңыр бояуы және т. б.
А. ОЖЖ: эпилепсия, шизофрения және т. б.
Б. Жүрек-қантамыр: ревматизм, атеросклероз және т. б.
В. Терілік: псориаз және т. б.
Г. Тыныс алу жүйесі: бронхиальды демікпе, аллергиялық альвеолит және т. б.
Д. Зәр шығару жүйесі: нефриттер, энурез және т. б.
- Хромосомалық аурулар
Адамның қалыпты соматикалық клеткаларының хромосомалық жиыны 46 хромосомадан тұрады (2n=46) . Әйел адамда 44 аутосомды және ХХ жұп хромосомасы болса, ер адамда 44 аутосомды және ХУ хромосомасы болады. Сәйкес келетін кариотипке құрылған формула келесідей үлгіде: 46, ХХ; 46, ХУ болады. Хромосомалық аурулар - хромосома құрылымы мен санының өзгеруіне байланысты дамитын туа біткен аурулардың өте үлкен тобы. Мұндай аурулар ата-ананың біреуінің жыныс клеткаларындағы мутацияның нәтижесінде туындайды. Ұрпақтан-ұрпаққа берілуі 3-5%. Бірақ хромосома жиынтығының өзгеруі қалыпты гаметадан түзілген зиготаның бірінші бөлінуі кезінде де болуы мүмкін. Өте жиі кездесетін хромосомалық ауру 21 хромосомамен тікелей байланысты. 21 хромосомадағы трисомия - балалардағы ақыл-есінің дамуының артта қалуымен көрінетін ерекше ауру түрі. Ең алғаш бұл ауру 1866 жылы ағылшын педиатры Даунның еңбектерінде сипатталды және содан кейін «Даун синдромы» деп аталды. Ал Н. А. Шерешевский 1925 жылы Х-хромосомадағы моносомияның алғашқы клиникалық сипаттамасын берді, одан кейін Г. Тернер 1938 жылы осы синдром туралы сипаттама берген болатын. Осы ауруды зерттеп анықтаған екі ғалымның атымен бұл ауру - Шерешевский - Тернер синдромы деп аталды. Хромосомалық аурулар барлық ұлттар мен этникалық топтарда таралған және кездесу жиілігі әрбір 1000 жаңа туған нәрестелердің арасында 7-8 кездеседі [6] .
Барлық хромосомалық ауруларды 2 топқа бөлуге болады:
а) Хромосома санының аномалиясымен байланысты. Бұл топқа 3 топша кіреді:
1) хромосома санының бұзылуы себебінен туындаған аурулар;
2) Х- және У- жыныс хромосомаларының саның артуы немесе кемуімен байланысты аурулар;
3) хромосоманың гаплоидты жиынтығының аздап қана артуымен байланысты аурулар.
б) Хромосоманың құрылымдық бұзылыстарымен байланысты.
Олардың негізгі себептері:
- Транслокация - гомологты емес хромосомалардың өзара орын ауыстыруы.
- Делеция - хромосома аймағының жоғалуы.
- Инверсия - хромосома бөлімдерінің 180° айналуы.
- Дупликация - хромосома бөлімдерінің екі еселенуі.
- Дөңгелек хромосоманың туындауы - хромосоманың екі иығындағы екі соңғы делецияның қосылуы.
Қазіргі таңда адамдарда хромосоманың бұзылыстарымен байланысты 700-ге тарта ауру түрлері белгілі. Оның 25% аутосомды трисомия, 46% жыныс хромосомаларының патологиясы болып табылады.
Хромосомалық аурулардың клиникалық көрінісі - ақыл-ес дамуының бұзылыстары мен туа біткен кемшіліктердің болуы [7] .
- Гендік аурулар
Гендік аурулар - гендердің өзгеруі нәтижесінде пайда болатын және клиникалық көріністері әртүрлі болып келетін аурулар. Мұндай аурулардың кездесу жиілігі 1:500 - 1:1 аралығында ауытқиды. Бір генде ауруды тасымалдаушы он не жүз мутацияның түрі болуы мүмкін. Гендік аурулардың тұқымқуалаушылық ерекшеліктері Г. Мендельдің заңымен анықталады. Мутация белок молекуласындағы сәйкес келетін полипептидті тізбек құрылымын бұза отырып, кез-келген генде туындауы мүмкін. Белок қызметінің өзгеруі әсерінен организмде биохимиялық өзгерістер туындап, арнайы клиникалық көрінісі бар тұқым қуалайтын ауруға алып келеді [3] .
Гендік аурулардың организмде туындауы келесідей сызбамен жүзеге асырылады: мутантты аллель → патологиялық бірінші өнім → кезектескен биохимиялық реакциялардың тізбегі → клеткалар → мүшелер → организм. Тұқым қуалайтын ауруды шақыратын мутация құрылымдық, транспорттық және эмбриональды белоктар мен ферменттерге әсер етеді. Бірінші туындаған гендік мутация фенотиптік көрініс береді, егер ген доминантты немесе ер адамның Х-хромосомасына орналасса, онда бұрын мұндай аурумен ауырмаған отбасыда аутосомды-доминантты немесе Х-тіркескен ауруы бар бала туылады. Рецессивті гендегі мутацияны анықтау өте қиын. Гендік аурулар көбінесе гетерезиготалы болып келеді [4] .
Гендік аурулардың кездесу жиілігі мен тұқымқуалау типі 2 - кестеде көрсетілген.
Кесте 2.
Гендік аурулардың таралуы [4] .
А-Д, А-Р,
Р-Х-тіркескен
Гендік аурулардың кездесу жиілігі 1000 жаңа туған нәрестелердің ортасында 2, 5-30% құрайды. Оның ішінде аутосомды-доминантты патология - 0, 2-0, 85% балаларда, аутосомды-рецессивті - 0, 2-0, 5% балаларда, ал Х-тіркескен - 0, 05-0, 7% құрайды. Гендік аурулар гетерогенді болып келеді. Мұндай гендердің гетерозиготалық жағдайы ұрпақтан-ұрпаққа фенотипке өз әсерін тигізбей-ақ беріле беруі мүмкін. Кейбір патологиялық гендерді қарағанда фенотиптік әсерімен байланысты Мендель-Моргандық ережеге бағынбауға болады. Мысалы, REТ-онкогеннің әртүрлі бөлігіндегі мутация клиникалық көрінісі жағынан әртүрлі болып келетін 4 тұқым қуалайтын ауруды тасымалдауы мүмкін. Олар: полиэндокринді аденоматоз ZA және ZB, отбасылық медуллярлы тереоидты карцинома, Гиршпрунг отбасылық ауруы.
Зат алмасудың көп санды аурулары, яғни көмірсу, ақуыз, май, билирубин, пурин алмасуының бұзылыстары гендік аурумен ауырған науқаста анық байқалады, яғни гендік аурулар негізінен зат алмасу үрдістерін бұзады [3, 6, 7] .
- Қалқанша безінің жалпы сипаттамасы
Қалқанша безі - адамның ішкі секреция бездерінің ішіндегі ең маңыздыларының, әрі ерте дамитын мүшелерінің бірі. Адамдарда қалқанша безінің бастамасы құрсақішілік даму кезеңінің 4-ші аптасының соңына қарай эндодерманың қалыңдауы арқылы жұтқыншақтың түп жағындағы бірінші және екінші жұп жұтқыншақ қалташығының арасында қос жақтаулы құрылымды, жұтқыншақпен байланысып тұратын төмпек болып түзіле бастайды. Бұл төмпек біріншілік фаренгиальды түтіктің эндодермальды қабатының (эндотелиальды) туындысы және 2-2, 5 мм шамасында жинақталған эпителиальды клеткалардың шоғыры болып табылады. Қалқанша безінің бастамасы 1-суретте көрсетілген.
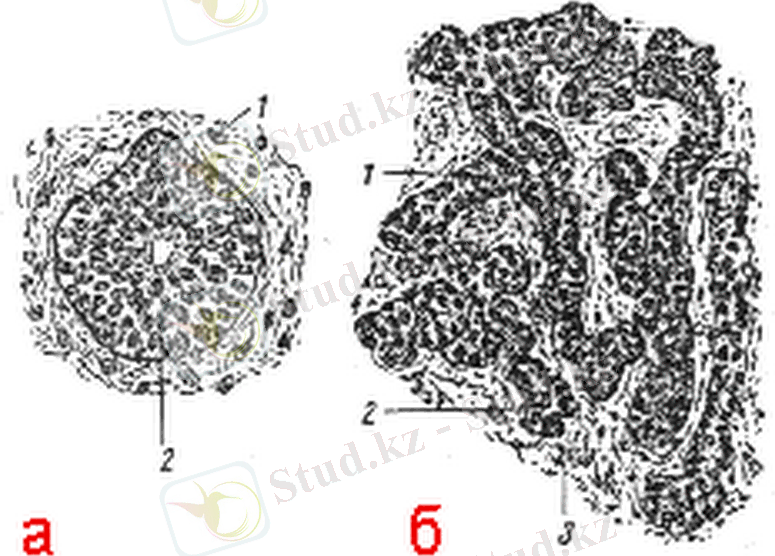
Сурет 1. Қалқанша безінің бастамасы. 1 - мезенхима, 2 - қалқанша безінің өсіндісі
Ары қарай өсінді екіге бөлінеді және каудальды бағытқа қарай көшеді, нәтижесінде эпителиальды тәж пайда болады. Дистальды бөлігі есі еселеніп, екі жақты құрылымды болады. Қалқанша безі гистологиялық жағынан толық дамуы әр әдебиеттерде әртүрлі көрсетілген. Оның проксимальды бөлігі жұтқыншақ пен тілдің соқыр өсіндісімен байланысып тұрады, ал қалқанша безімен дистальды байланыс пирамидалық үлес түрінде сақталады. Бездің екі жағының үлесі жұтқыншақ төмпек қалташығынан дамиды. Қалқанша безінің бастамасының дамуы эмбрионалды дамудың соңғы апталарында өтеді және ол жүректің және қолқа тамырының алғашқы белгілерімен өте тығыз байланыста болады. Бұл кезеңде қалқанша безінің бастамасының қанмен қамтамасыз етілуін фарингиальды артерия қамтамасыз етеді, ол қолқа қалташығынан шығып, мезенхималық ұлпаның айналасында магистральды артерияға айналады. 2-ші айдың басында оның жоғарғы бөлігі жұқара бастайды, соңында семіп қалады. Ал проксимальды бөлігінен тек тіл түбірімен жалғасқан саңылау ғана қалады. Үлестерінің бір-бірімен қосылуы жатырішілік дамудың 7-ші аптасында өтеді. Алдымен қалқанша безінің белгісі (дивертул) эпителиальды клеткалардың көптеген санынан құралады, ары қарай бөлінгіш клеткалар тәж байланысы трабекуланы түзеді. Содан кейін эпителиальды трабекулалар жылдам өсіп, соңында одан фолликулалар түзіледі. Қалқанша безінің құрылымдарының бастамалары 2-суретте көрсетілген.
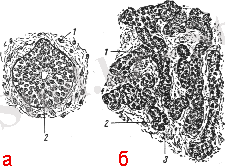
Сурет 2. Қалқанша безінің бастамалары. 1 - эпителиальды тәж, 2 - фолликула бастамасы, 3- мезенхима
Фолликулалар арасындағы аралықтарда жүйке және қантамырлармен қамтамасыз етілген мезенхима өседі [8-10] .
Қалқанша безінің бастамасындағы жақсы жетілген ұсақ фолликулалар жатырішілік дамудың 2-ші айының аяғы мен 3-ші айдың басында анық бола бастайды. Түзілген фолликулалар коллоидтан тұрады және ол йодты жинақтауға қабілетті болып келеді.
Ұрықта қалқанша безінің дамуы (жүктіліктің 80 күнінен туылғанға дейін) үш кезеңнен өтеді: коллоид алды (тиреоглобулин алғаш синтезделетін біріншілік безде экстрамедуллярлы коллоидты кеңістік түзіледі), ерте коллоидты (йодтың белсенді сіңірілуімен және тиреоидты гормон мен коллоид түзілуімен сипатталады) және фолликулярлы (фолликулдардың дамуы және олардың коллоидтармен толығуы) [10-12] .
Йодпероксидазаның белсенділігі жатырішілік дамудың 4-5-ші айларында жоғарылай бастайды. Сонымен, жүктіліктің 11-ші аптасының соңы мен 12-ші аптасының басында ұрықтың қалқанша безінің йодты жинақтауға, тиреоидты гормонды өндіруге және синтездеуге қабілетті болады. Сонымен қатар, қалқанша безінің клеткалары 11-аптада моно- және дийодтирозинді, тіпті тироксинді анықтай бастайды [9, 10] .
3-4 айда фолликула коллоидтары сіңіргіш көпіршіктердің көрінуімен ерекшеленеді, йод органикалық қосылыстармен қосылғаннан бастап клеткалардың негізгі қызметі басталады, яғни, без қызметтік жағынан жетіледі [12] .
Гестацияның ерте кезеңінде ұрықта қалқанша безінің қалыптаса бастауы ананың аналогты эндокринді жүйесіне оның қызметінің тәуелсіз болуымен көрінеді. Ұрықжолдастық тосқауыл анадан құрсақтағы балаға гипоталамо-гипофизарлы жүйенің гормондарының енуін тежейді, бірақ сол уақытта белгісіз бір мөлшерде бос тироксин (Т4) және трийодтиронин (Т3) ұрыққа және ұрық жанындағы суға енеді. Тироидты гормонға деген ұрықжолдастық тосқауылдық өткізгіштік деңгейі жүктілік мерзіміне тікелей байланысты. Бірінші триместрде ұрыққа бос тироидты гормондар салыстырмалы түрде өте көп мөлшерде енеді, ал екінші және үшінші триместрде фето-ұрықжолдастық тосқауылдың түзіле бастауынан және ұрықтың өз қалқанша безінің қызметі қалыптасуынан анадан ұрыққа тироидты гормондардың өтуі және оны пайдалану минимальды деңгейге дейін төмендейді. Бұл кезеңде ана мен баланың қалқанша безінің гормондық қызметінде физиологиялық автономия қалыптаса бастайды. Бірақ ана қанында Т3 және Т4 концентрациясы патологиялық жоғары болса, онда бұл гормондар ұрықжолдас арқылы өтуі мүмкін. Сонымен қатар, тиротұрақтандырушы препараттарды жүктілік кезінде тиротоксикозды емдеуге қолдану гормонның ұрықжолдас арқылы бос өтуін және ұрықтың қалқанша безінің қызметін нашарлататынын есте сақтаған жөн [12] .
Жүктіліктің екінші жартысында қалқанша безі өзіндік қызметке ие болады, яғни, тироксин бөле бастайды. Одан ары сандық өзгерістер орын алады: фолликулалардың жаңа түрлері пайда болады және көлемдері жағынан үлкейіп, метоболиттік белсенділігі артады [13] .
Электронды-микроскоптық зерттеу кезінде 4-айлық эмбрионның тиреоцитінің құрылысы ересек адамның безінің құрылысымен бірдей болған. Нәрестенің қалқанша безі қызметтік жағынан жетілген және салмағы 1 г болып келеді. Нәрестелерде, балаларда және ересектерде қалқанша безінің салмағы салыстырмалы түрде бірдей болып келеді [14-16] .
Нәресте туылғаннан кейін, ерте неонатальды кезеңде, гипофизарлы-тироидты жүйеде гормондардың концентрациясында айтарлықтай өзгерістер орын алады. Кіндік қанында Т4 мөлшері ана қанымен салыстырғанда 1, 2 есе аз болады. Туылғаннан кейінгі алғашқы екі тәулікте Т4 деңгейі шамалы ғана жоғарылайды, ал үшінші тәулікте ең жоғары деңгейге дейін жетеді де, содан кейін екі-үш апта көлемінде қайтадан ақырын төмендеп белгілі бір мөлшерге жетеді. Ал кіндік қанында Т3 концентрациясы ана қанымен салыстырғанда бес есе төмен болады. Туылғаннан кейінгі бірінші тәулікте Т3 мөлшері ең жоғарғы деңгейге дейін жетеді де, кіндік қанындағы гормонның жинақталуы 3-4 есе жоғарылайды, ал сосын үшінші тәулікте Т3 мөлшері төмендей бастайды. Екінші реттік гормон деңгейінің жоғарылауы нәресте туылғаннан төртінші аптаға дейін созылады. Ерте неонатальды кезеңде бейімделудің өзгергіштігіне ТТГ (тиретропты гормон) концентрациясы ұшырайды. Нәресте кіндігіндегі ТТГ концентрациясы анасындағы гормон концентрациясынан 4 есе жоғары болады. Нәресте туылғаннан кейінгі 30 минуттан соң ол алты есеге жоғарылайды, тек бірінші тәуліктің соңына қарай минимальды деңгейге дейін төмендеп кетеді. Келесі 3-6 тәулікте қандағы ТТГ концентрациясы ақырын жоғарылайды да, гормон деңгейі тұрақтана бастайды. Кіндіктегі тироксин байланыстырушы глобулин мөлшері анасымен салыстырғанда 1, 5 есеге төмен болады. Туылғаннан кейінгі бірінші тәулікте тироксин байланыстырушы глобулин мөлшері бірнеше рет жоғарылайды да, үшінші тәулікте төмендей бастайды, ал сосын қайта жоғарылап қалыпты мөлшерге жетіп, тұрақтанады [12, 13] .
- Қалқанша безінің гистогенезі және оны реттеуші гендер
Қалқанша безінің құрылымдық-қызметтік бірлігі - фолликулалар. Олар қоршалған шартәрізді немесе ішкі қуысы аздап созылыңқы болып келеді. Көлемі 20-300 мкм аралығында болады. Қалқанша безі гистологиялық құрылысы жағынан дәнекер ұлпасынан тұрады, қабаттары тереңге еніп, оны екі бөлікке бөледі. Фолликула қуысында күрделі белокты заттар - эпителиальды немесе А-клеткалар (тиреоциттер) өндіретін коллоид болады. А-клеткамен қатар, қалқанша безінің ұлпасында С-клеткалар (парафолликулярлы) болады. Олар фолликулалар арасында орналасады. Бұл клеткалар организмдегі кальций және фосфор айналымын реттеуші негізгі гормондардың бірі кальцитонинді өндіреді. Тиреоциттер- бірқабатты кубты эпителийлі безді клеткалар. Ұшында микротүтікшелері бар, ал көрші клеткалары бір-бірімен тығыз байланысқан көпсанды тосқауылдар - десмосомалар болып табылады [17, 18] .
Қалқанша безінің фолликуласында тиреоидты гормондар тироглобулинмен байланысқан, ол синтез жүретін негізгі матрица болып табылады. Тироглобулин тироидты гормондарды негізгі сақтаушы, әрі протеолиз нәтижесінде қанға түсуін және олардың босатылуын жүзеге асырушы болып табылады. Қанда тироидты гормондар сарысулы белоктық кешен түрінде айналымда жүреді, бірақ физиологиялық белсенділігі жағынан тироидты гормон фракциясының белоктарымен байланыспаған бос күйінде болады. Т4 және Т3 гормондарының өз рецепторлары бар, олар шеткі мүшелермен тироидты гормондарды байланыстыруға арнайылық қасиет көрсетеді және олардың биологиялық әсерлерінің жүзеге асырылуымен қамтамасыз етуші рөл атқарады [12] .
... жалғасыСіз бұл жұмысты біздің қосымшамыз арқылы толығымен тегін көре аласыз.
- Іс жүргізу
- Автоматтандыру, Техника
- Алғашқы әскери дайындық
- Астрономия
- Ауыл шаруашылығы
- Банк ісі
- Бизнесті бағалау
- Биология
- Бухгалтерлік іс
- Валеология
- Ветеринария
- География
- Геология, Геофизика, Геодезия
- Дін
- Ет, сүт, шарап өнімдері
- Жалпы тарих
- Жер кадастрі, Жылжымайтын мүлік
- Журналистика
- Информатика
- Кеден ісі
- Маркетинг
- Математика, Геометрия
- Медицина
- Мемлекеттік басқару
- Менеджмент
- Мұнай, Газ
- Мұрағат ісі
- Мәдениеттану
- ОБЖ (Основы безопасности жизнедеятельности)
- Педагогика
- Полиграфия
- Психология
- Салық
- Саясаттану
- Сақтандыру
- Сертификаттау, стандарттау
- Социология, Демография
- Спорт
- Статистика
- Тілтану, Филология
- Тарихи тұлғалар
- Тау-кен ісі
- Транспорт
- Туризм
- Физика
- Философия
- Халықаралық қатынастар
- Химия
- Экология, Қоршаған ортаны қорғау
- Экономика
- Экономикалық география
- Электротехника
- Қазақстан тарихы
- Қаржы
- Құрылыс
- Құқық, Криминалистика
- Әдебиет
- Өнер, музыка
- Өнеркәсіп, Өндіріс
Қазақ тілінде жазылған рефераттар, курстық жұмыстар, дипломдық жұмыстар бойынша біздің қор #1 болып табылады.

Ақпарат
Қосымша
Email: info@stud.kz